

Dispersions
The International Union of Pure and Applied Chemistry (IUPAC) definition of dispersion is a material comprising more than one phase where at least one of the phases consists of finely divided phase domains, often in the colloidal size range, dispersed throughout a continuous phase (Slomkowski et al., 2011). Dispersions are sub-classified based on the size of the discontinuous phase particles, whether or not precipitation occurs, and the presence of Brownian motion. Small particles that do not sediment and are kept apart via Brownian motion are termed colloids varying between 5 nm and 0.5 µm, while particles >0.5 µm will eventually sediment and are termed suspensions. No finite size differentiates colloids from suspensions because the interface characteristics are equally important as both parameters alter stability, which ultimately classifies stable dispersions as colloids and non-stable dispersions as suspensions. Suspensions are typically opaque as the particles exceed the wavelength of light, causing light to reflect. The Tyndall effect is a simple way to differentiate transparent solutions from transparent colloids; passing a light or laser beam through a solution cannot be observed; however, the presence of a colloid, such as diluted skim milk (e.g., casein micelle) or a flour suspension, will make the light beam apparent as it passes through the dispersion. Colloids are essential and relevant everywhere in biological fluids, foods, and delivery systems for fertilizer and pharmaceuticals.
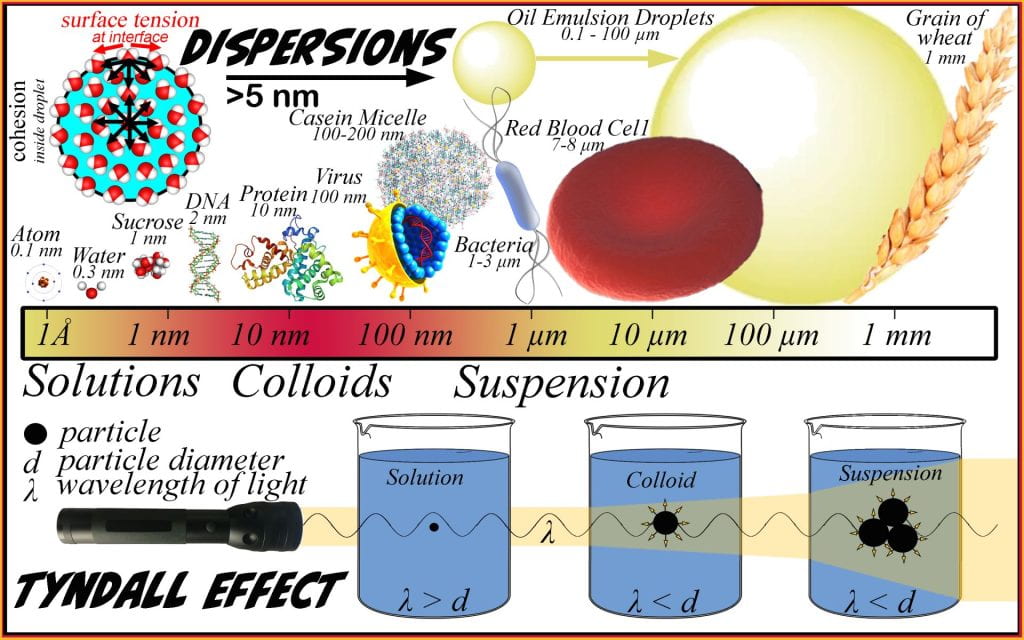
A key differentiating factor between solutions and dispersions is the presence of an interface between incompatible phases. When the dispersed and continuous phases are incompatible (i.e., oil and water), an interface gives rise to unbalanced cohesive forces generating a surface tension that has a net attraction toward the droplet, minimizing the radius and surface area resulting in spherical droplets. Thermodynamics will minimize the unbalanced force generated across the interface when a surface is flexible, such as an oil-in-water emulsion or a soap bubble-in-air. In a two-phase oil-in-water system, the surface tension is constant, so the force can only be lowered by decreasing the surface area that the surface tension can act across. Spheres form because they are at the maximum volume-to-surface area ratio; the other feature to arise due to the surface curvature is the Laplace pressure. The Laplace pressure is the outward force exerted at the droplet interface. Practically, this is obvious if considering blowing up a balloon; as air is blown into the balloon, the pressure increase inside but not outside the balloon and when more air is blown in, the internal pressure increases more. If Laplace pressure does not result or the pressure inside is less than outside, the surface will become undulated and puckered, which does not occur. Dispersions are commonly classified depending on the state (e.g., solid, liquid, gas) of continuous and discontinuous phases.

A continuous solid phase and discontinuous gas phase form solid foams such as bread, meringues, and cookies, while a liquid dispersed phase in a solid forms gels, such as cheese, jello and jams. Dispersions containing solids in the dispersed and continuous phases are termed solid solutions, including ceramics and metal alloys. A continuous liquid phase with the dispersed phase as air forms foams (whipped cream), while liquids form emulsions (milk and salad dressings) and solids form suspensions (ice cream). Although few foods exist with a continuous gaseous phase, they still make up an important class of dispersed systems such as vapors (gas-in-gas), fog (liquid-in-gas) and smoke (solid-in-gas).
Sedimentation and Creaming

A particle suspended in solution must displace an equal volume continuous phase. When the density of the particle is less than the continuous phase, creaming occurs (e.g., less dense particles rise in solution); conversely, when the particle density exceeds the continuous phase density, sedimentation occurs (more dense particles fall out of solution). The rate of creaming and sedimentation are determined using Stoke’s law which combines the downward gravitational force counteracted by the buoyant force of the displaced continuous phase and the drag force. The separation of particles accelerates until the drag force adjusts itself to make the net force on the particle equal to zero, after which the spherical particle travels at a constant terminal velocity (υs).
The gravitational force is the particle mass, calculated from the particle volume (Vparticle = 4/3πR3), where R is the radius, multiplied by the density of the particle (r2), and gravitation force (~ 9.81 ms-2). The buoyant force is identical to the gravitational force, except it replaces the particle density r2 with that of the continuous phase r1; hence it considers a sphere of the displaced continuous phase. Once the spherical particle is in motion, a drag force (F=6πµRυs) is generated, acting in the opposite direction the particle travels and dependent on the continuous phase viscosity (µ).
Stoke’s law calculates the terminal velocity that the particle will separate, which is directly proportional to the difference in density between the particle and continuous phase, a positive squared correlation to particle radius, and inversely proportional to the viscosity. Separation accelerates when the density difference or particle radius increases and continuous phase viscosity decreases. Stokes’ law is particularly useful for homogenously sized particles, but when the dispersion contains a range in particle sizes, non-uniform velocities lead to particles separating at different speeds. Large particles travel faster than small particles, which causes sweeping flocculation where particle aggregate due to different velocity profiles, and as particles flocculate, their effective radius increases, further accelerating separation. For oil-in-water emulsions, stability is increased when the density of the oil phase is increased closer to the density of water, accomplished by brominating vegetable oil, increasing the continuous phase viscosity by adding a low concentration of protein or polysaccharides, and decreasing the particle size, by increasing the power of homogenization.

Ostwald’s Ripening

Beyond nanodispersions, particles are not thermodynamically stable, therefor kinetic barriers to separation must be applied to maintain the desired structure until the best-before date. As stated above, the interface of the spherical droplet has unbalanced cohesive forces giving rise to surface tension. The system attempts to reach equilibrium by forming the smallest surface area for the surface tension to act. Thus, the internal pressure is inversely proportional to the radius and positively proportional to surface tension. The internal pressure, or Laplace pressure, acts outward, ensuring a smooth surface. Small droplets have a higher internal pressure than large droplets, and the solubility of the dispersed phase is proportional to this pressure. Consequently, molecules dynamically exchanging at the interface over time favor the migration of molecules from smaller droplets to larger ones without the droplets coming into contact with each other. Over time, this causes emulsions to coarsen, where large drops, particles, and crystals grow at the expense of small droplets, which shrink and eventually disappear.
Ostwald’s ripening cannot be completely prevented but can be slowed by adding amphiphilic molecules to the interface lowering the surface tension, or increasing the continuous phase viscosity, especially near the interface, to impede the diffusion of newly soluble molecules away from the small crystals.
Stabilization and Destabilization of Dispersions
Particles in dispersions are constantly in motion and can diffuse through the continuous phase causing sedimentation or creaming depending on the density difference between the discontinuous and continuous phases. Increasing the viscosity of the continuous phase or minimizing the density difference between phases slows the separation rate. Ostwald’s ripening arises due to the higher internal pressure of small droplets than larger droplets, causing higher solubilization of molecules in the small droplets. Over time, this causes large particles to grow at the expense of smaller droplets which eventually completely disappear. Flocculation and coalescences occur when two molecules come into contact with each other; coalescence is non-reversible because, upon contact, they combine into one spherical droplet which a single shared interface.
Flocculation is reversible; when droplets come into contact, the interfacial layer is not displaced, and discrete particles remain clumped. Partial coalescence is when particles come into contact, and a small interface area is shared between the two droplets. Partial coalescence is common when the dispersed phase is partly crystalline, and the crystals puncture the interface and reside across both droplets. Partial coalescence is essential in forming the desired structure of whipped and ice creams. Finally, phase inversion occurs when the concentration of the dispersed phase is similar to the continuous phase, which undergoes spontaneous inversion, causing an oil-in-water emulsion to transition to a water-in-oil emulsion. Phase inversion is temperature dependent and greatly affected by the molecules added to stabilize the interface.
Dispersion Interfaces and Amphiphilic Molecules
While the interface of the dispersion has a small effect on sedimentation and creaming, adding molecules to the interface can effectively increase the density of the discontinuous phase, and if it becomes closer to the dispersed phase, separation is slowed, as apparent in Stoke’s law. However, the chemistry of the interface is also an important parameter that alters the net attractive and repulsive forces between droplets and minimizes the interfacial surface tension (lowers the rate of Ostwald’s ripening) and can prevent phase inversion. The cohesion, or attractive force, for water, is dominated by strong hydrogen bonds, while oils interact mainly by weak van der Waals interface arising from the long hydrocarbon chains. Both oil and water have unbalanced forces at the interface; for oil, it is van der Waals interactions, and for water, it is hydrogen bonding, both of which are incompatible. These unbalanced forces give rise to surface tension; the oil-water interfacial tension is 35 mN/m, and for air-water, it increases to 72 mN/m. Amphiphilic molecules contain hydrophobic and hydrophilic ends and are added to dispersions to decrease the magnitude of these unbalanced forces. Adding amphiphilic molecules (surfactants) to a dispersed system significantly decreases the system’s surface tension, slowing Ostwald’s ripening.
Emulsifiers or surfactants are small or polymeric molecules added to dispersions to reduce the interfacial tension. These include proteins, which unfold, exposing hydrophobic amino acids to the oil or non-polar phase and hydrophilic amino acids to the water or polar phase. Also, small molecules such as fatty acids (hydrocarbon chain attached to a carboxylic acid), monoglycerides, phospholipids, sorbitan fatty acid esters (Spans) and polyoxyethylene sorbitan fatty acid esters (Tweens) can effectively align their polar portion into the water phase and non-polar side to the oil phase. Both polymeric and monomeric emulsifiers significantly reduce the system’s unbalanced interface forces. Dispersed systems are tailored depending on the compositions and concentration of the dispersed and continuous phases by selecting an appropriate surface active (amphiphilic) molecule (emulsifier/surfactant). Amphiphilic molecules are characterized and selected based on their hydrophilic-lipophilic balance (HLB), which balances the size and strength of a molecule’s hydrophilic and lipophilic moieties. HLB values numerically express the polar portion’s size and strength relative to the molecule’s non-polar portion. HLB is calculated using an arbitrary scale of 0-20 and calculated simply from the fraction of the molecular weight of the hydrophilic groups (Mh) to the molecular weight of the molecule (M).
HLB = 20 X Mh/M

HLB values are essential to select the correct amphiphilic molecule for the dispersion. For foods with continuous water phases and dispersed oil, the HLB value should be between 8-16; while for continuous oil phases, the HLB value should be between 3-6. Selecting the incorrect HLB value often leads to phase inversion. Food-grade emulsifiers are commercially available for every HLB value, providing flexibility when designing stable dispersed systems. For example, Spans have low HLB values ranging from 1.8 to 8.6; while Tweens are more hydrophilic, having higher HLB values ranging from 9.6 to 16.7. In addition to lowering the system’s interfacial tension, emulsifiers can impart other stabilizing factors, including electrostatic and stearic repulsion.

DLVO Theory and the Gibbs-Marangoni-Effect

If the interface contains an ionizable emulsifier (e.g., sodium lauryl sulfate) at specific pH values, the groups will be either deprotonated or protonated and carry a negative or positive charge leading to electrostatic repulsion. In the case of a negatively charged interface, positively charged ions and molecules, which are soluble in the continuous phase, are attracted to the droplet surface, creating the stern layer that is the opposite charge carried by the interface. After the stern layer is the double layer over which the remaining charge is neutralized and beyond which no electrostatic repulsion exists.
When dispersed in a water phase, nonpolar-oil droplets, coated in ionized emulsifiers, are drawn to each other by van der Waals attraction (VdW), weak forces that act over large separation distances between particles; as particles come closer together, the VdW interactions increase. Simultaneously, when the distance between droplets decreases to the size of the double layer, weak electrostatic repulsion forces begin to act on the particles. At this point, the net VdW attraction exceeds the electrostatic repulsion, and the particles continue to move closer until they reach a distance equal to their stern layer. When only the stern layers separate particles, the electrostatic repulsion exceeds the VdW interactions, pushing the particles apart.
This phenomenon is the basis of the DLVO (Derjaguin, B., Landau, L., Verwey, E., Overbeek, T.) theory used to explain the aggregation of aqueous dispersions describing the attractive and repulsive force between charged surfaces that interact through a continuous liquid phase. If the particle reaches a distance less than the stern layer, irreversible aggregation leads to flocculation and coalescences.
Another stabilizing factor imparted by emulsifiers occurs when the amphiphilic molecules extend outward from the droplet interface into the continuous phase but do not carry an ionic charge. The Gibbs-Marangoni effect is the mass transfer along an interface between two phases due to a surface tension gradient. The longer, more flexible the emulsifier segment that extends into the aqueous phase, the greater the Gibbs-Marangoni effect. As two droplets approach, the extended segments encroach on each other, and the surface becomes undulated or uneven, increasing the surface area, and in response, emulsifiers flow to equilibrate the surface tension evenly across the undulated droplet. Next, the cascade has a concentration or osmotic gradient between the bulk dispersed phase and the dispersed phase between particles. This causes the inward flow of water to equilibrate the osmotic gradient pushing the particles apart. As the particles move apart, the spherical shape is restored, and emulsifiers redistribute evenly across the surface area, reestablishing equilibrium

Works cited
Derjaguin, B.; Landau, L. 1941. Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solutions of electrolytes. Acta Physico Chimica URSS, 14: 633.
Verwey, E. J. W.; Overbeek, J. Th. G. 1948. Theory of the stability of lyophobic colloids. The Journal of Physical and Colloid Chemistry. 51: 631–636,
Slomkowski, S. et al., 2011. Terminology of polymers and polymerization processes in dispersed systems (IUPAC Recommendations 2011). Pure and Applied Chemistry. 83: 2229–2259.